


Календарь новостей
| « Апрель 2024 » | ||||||
|---|---|---|---|---|---|---|
| Пн | Вт | Ср | Чт | Пт | Сб | Вс |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| 8 | 9 | 10 | 11 | 12 | 13 | 14 |
| 15 | 16 | 17 | 18 | 19 | 20 | 21 |
| 22 | 23 | 24 | 25 | 26 | 27 | 28 |
| 29 | 30 | |||||

Обмен ссылками
Эпидермодисплазия верруциформная Левандовского—Лютца. Множественная самоизлечивающаяся плоскоклеточная эпителиома Фергюссон—Смита
21.01.11, посмотрело: 5 460
Эпидермодисплазия верруциформная Левандовского—Лютца (ЭВЛЛ) (MIM 3033350, 226400) — редкое, обычно аутосомно-рецессивно наследуемое заболевание с уникальной предрасположенностью к инфицированию кожи специфическими разновидностями ВПЧ. Проявляется в раннем детстве. У больных ЭВЛЛ выделено несколько типов ВПЧ, которые можно разделить на две группы:
1) с высоким онкогенным потенциалом — 5, 8 и 47-й типы, которые обнаруживаются в 90% плоскоклеточных раков кожи, развившихся на фоне очагов ЭВЛЛ;
2) с низким злокачественным потенциалом — 3, 14, 20, 21 и 25-й типы, которые обычно обнаруживаются в доброкачественных очагах ЭВЛЛ.
Генетические дефекты и наличие ВПЧ (чаще 5-го типа) у больных ЭВЛЛ обычно ассоциируются с нарушением клеточного звена иммунитета (подавление иммунного ответа и нарушение клеточно-опосредованной цитотоксичности, хотя антигенпрезентирирующая активность клеток Лангерганса не нарушена) и воздействием таких канцерогенных факторов, как ультрафиолетовое излучение спектра В и ионизирующая радиация.
Клинически верруциформная эпидермодисплазия Левандовского—Лютца характеризуется множеством пятен и бородавок, главным образом плоского типа, которые имеют тенденцию к слиянию и распространяются иногда на всю поверхность предплечий, голеней, лица. В отдельных случаях формируются участки ограниченного гиперкератоза, дисхромии, алопеции. У 30-50% больных ЭВЛЛ, особенно на открытых участках кожи, развиваются болезнь Боуэна, бовеноидный папулез с трансформацией в плоскоклеточный рак в 20-30% случаев, реже базалиомы.
Первый очаг плоскоклеточного рака кожи возникает в среднем в 27 лет, то есть почти на 40 лет раньше, чем в обшей популяции. Развитие плоскоклеточного рака сопровождается усилением роста бородавчатоподобных элементов, слиянием их в крупные инфильтрированные бляшки, которые приобретают красный или красно-коричневый цвет, изъязвлением. Плоскоклеточные раки, развивающиеся при ЭВЛЛ. обычно характеризуются периневральной инвазией и частым метастазированием в лимфатические узлы, а иногда и во внутренние органы, что приводит к летальному исходу. Средний возраст больных на момент развития метастазов — 34 года. Следует учитывать, что агрессивному клиническому течению рака кожи у больных ЭВЛЛ способствует рентгенотерапия.
Лечение рака кожи при верруциформной эпидермодисплазии Левандовского—Лютца проводится с использованием химиотерапевтических препаратов (проспидин, блеомицин) в сочетании с хирургическим удалением опухоли. Эффективны ароматические ретиноиды. Бородавчатоподобные элементы удаляют методами криодеструкции, электрокоагуляции, с помощью неодимового лазера; наружно назначают 30-50% проспидиновую, 5% флуороурациловуто мази.
Профилактика злокачественной трансформации верруциформной эпидермодисплазии Левандовского—Лютца заключается в использовании солнцезащитных средств. Учитывая важную роль в развитии озлокачествления ВПЧ-5, целесообразно повышение иммунологической реактивности организма как больных, так и их ближайших родственников с использованием препаратов интерферона. Желательно также медико-генетическое консультирование больных и их ближайших родственников перед вступлением в брак.
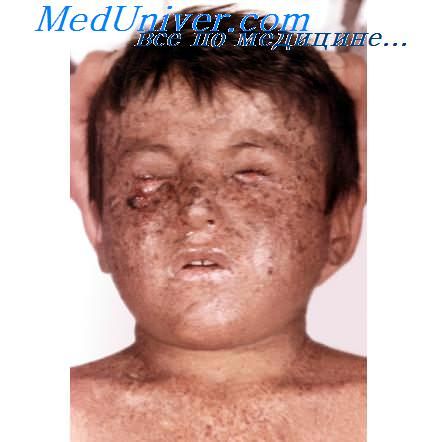
Множественная самоизлечивающаяся плоскоклеточная эпителиома Фергюссон—Смита
Множественная самоизлечивающаяся плоскоклеточная эпителиома Фергюссон—Смита (ТУ1СПЭФС) (MIM 132 800) - редкое наследственное заболевание, тип наследования аутосомно-доминантный. Предполагается, что ген, определяющий развитие заболевания, картируется в 9-й хромосоме (q22-q31). Впервые описана в двух шотландских семьях Дж. Фергюссон-Смитом.
При множественной самоизлечивающей плоскоклеточной эпителиоме Фергюссон—Смита происходит быстрое развитие кератоакантомаподобных узлов на лице и других облучаемых солнцем участках кожи. Хотя возраст больных варьирует от 8 до 62 лет, чаще заболевание встречается в подростковом или юношеском возрасте. Кожные опухоли представлены множественными куполообразными узлами с центральным кератиновым кратером, которые в течение нескольких недель увеличиваются, изъязвляются и подвергаются спонтанной инволюции с образованием вдавленных рубчиков неправильной формы. Если клинически элементы напоминают кератоакантому, то по гистологическому строению они нередко неотличимы от плоскоклеточного рака кожи. Изредка они метастазируют.
Синдром Ротмунда—Томсона
Синдром Ротмунда—Томсона (СРТ) (син.: врожденная пойкилодермия) — редкий наследственный синдром, тип передачи аутосомно-рецессивный (MIM 268 400). Несколько чаще отмечается у женщин (1,4:1). Генетические и биохимические дефекты при СРТ не определены, но отмечено снижение репарации ДНК в культивированных фибробластах после УФ-С и гамма-облучения. Наличие хромосомной нестабильности предполагается, исходя из высокой частоты ассоциации синдрома со злокачественными новообразованиями.
Кожные поражения синдрома Ротмунда—Томсона возникают в первые 3-6 мес и проявляются диффузной эритемой, иногда отеком и везикуляцией открытых участков кожи: лица, ушных раковин, тыла кистей, разгибательных поверхностей конечностей. Позже на этих же местах возникает пойкилодермия (телеангиэктазии, точечные участки гипо- и гиперпигментации, атрофии), напоминающая проявления хронического радиационного дерматита. В дальнейшем высыпания могут распространяться на ягодицы, шею, туловище. Прогрессирование этих поражений продолжается до возраста 3-5 лет. На втором десятилетии жизни у больных развиваются кератотические и бородавчатые высыпания на кистях, предплечьях, стопах и толенях. С возрастом фоточувствительность уменьшается, но сформировавшиеся в детстве высыпания персистируют в течение всей жизни. Для СРТ также характерны ювенильная катаракта, дистрофия ногтей и волос (разрежение или отсутствие волос на голове, бровей, ресниц, волос на лобке, в подмышечных впадинах), нарушение потоотделения, гипоплазия зубов с высокой частотой кариеса, нарушения физического развития, костные уродства, эндокринные расстройства, гипогонадизм в сочетании с низким ростом и т.д.
Синдром Ротмунда—Томсона ассоциируется с развитием злокачественных новообразований внутренних органов костей, крови (остеосаркома, рак желудка, лейкозы и т.д.) и немеланотических раков кожи: множественных очагов болезни Боуэна, плоскоклеточного рака кожи, базалиомы, возникающих на фоне очагов типеркератоза и атрофии.
Гистологически у детей при синдроме Ротмунда—Томсона обнаруживают уплощение и атрофию эпидермиса с отеком дермо-эпидермального соединения, иногда — некоторое расширение сосудов дермы и их периваскулярную инфильтрацию лимфоцитами; на открытых участках кожи взрослых — фрагментацию эластических волокон дермы в сочетании с пятнистым бовеноидным дискератозом в эпидермисе.
Дифференциальный диагноз синдрома Ротмунда—Томсона проводится с синдромом Коккейна, синдромом Вернера, врожденным дискератозом, прогерией, гипогидротической эктодермальной дисплазией, синдромом Елума.
Лечение синдрома Ротмунда—Томсона гиперкератотических, бородавчатых высыпаний и рака кожи проводится хирургически, с помощью криодеструкции, лучей лазера, используют также мази с цитостатическими препаратами (50% проспидиновую, 5% флюороурациловую), прием внутрь ароматических ретиноидов. Для лечения телеангаэктазии в области лица применяют аргоновый лазер. Важное значение имеет защита кожи от солнечного облучения. Лица пожилого возраста должны консультироваться дерматоонкологом с целью раннего выявления злокачественных опухолей кожи.
По материалам: